Read Secondary Schizophrenia Online
Authors: Perminder S. Sachdev
Secondary Schizophrenia (115 page)
ants associated with disease
[63, 64, 65];
and (ii) the
ants underlying many Mendelian disorders. Another
Chapter 23 – The status of genetic investigations of schizophrenia
impetus for employing linkage in psychiatric genetics
gree that has undergone thousands of meioses (gen-is that it does not require a priori knowledge about
erations) and displays LD (usually within distances
the location of disease genes. Linkage has been widely
< 0.2 cM) between an associated marker allele and the
applied for genomewide screening – typically using
untyped causative allele(s). Thus, association studies
several hundred STRP markers (and more recently,
appear to be more powerful than linkage studies for
panels of several thousand SNP markers) of known
identifying common genetic variants of small to mod-location. The goal of such studies is to identify one or
est effect on overall disease risk
[19,
70, 79].
The power
more chromosomal regions showing convincing evi-of a primary association study is influenced by sam-dence of linkage to SZ (criteria provided later). Sub-ple size, true effect size of the disease-causing allele,
stantial sample sizes are required to detect variants of
disease allele versus control allele frequency, and the
modest effect
[70, 75]
and the identified region(s) are
extent of LD between marker and disease allele
[80].
often very large, that is, > 20cM [20 million base pairs].
To detect variants of small effect (odds ratio: 1.3–1.5),
Follow-up studies with a higher density of markers
similar to those anticipated in SZ, thousands of cases
may then be conducted to extract maximal linkage
and controls would be required
[70, 81].
information from the implicated region(s) and suf-Association analysis is conducted in either case-
ficiently constrain the region to allow fine-scale LD
control samples or nuclear families
[19,
82].
In case-mapping (discussed later).
control studies, the presence of population stratifica-Stimulated by the feasibility of genomewide link-tion – arising when case and control groups differ in
age scans of pedigree cohorts, analytic methods were
the prevalence of distinct ethnic groups – can increase
developed to meet the need for (i) multipoint linkage
the type I error rate, as any trait more common in one
analysis with many markers across the genome; and
group will show association with alleles having higher
(ii) robustness in the face of uncertainty about mode
frequency in that same group
[83]
. Careful selection of
of inheritance in non-Mendelian diseases. Guidelines
cases and controls from relatively homogeneous popu-for the interpretation of results have been another
lations, especially prospective cohorts, can reduce this
critical development. Specific statistical criteria have
bias
[84]
as can the use of analytic methods that cor-been proposed
[76],
based on the number of times
rect for population structure
[85, 86, 87,
88].
Family-one would expect a result by chance in a genome-based methods such as the transmission disequilib-wide scan (GWS). Standard thresholds are suggestive,
rium test (TDT) are robust to population stratification,
significant, and highly significant linkage, which are
as control alleles are determined from family members
expected to occur by chance once, 0.05 times, and
(usually parents) of the affected individual
[89].
The
0.001 times, respectively, in a GWS. For sib pair stud-use of trios (both parents plus affected offspring) also
ies, these categories correspond to pointwise signifi-increases the accuracy of haplotype inference
[90].
cance levels of 7
×
10
−
4, 2
×
10
−
5, and 3
×
10
−
7, and
Association studies have typically been used for
LOD scores of 2.2, 3.6, and 5.4. It has been widely
evaluating defined candidate regions and/or disease
suggested that these guidelines are overly stringent,
susceptibility genes. However, the public availability of
due to potentially unrealistic assumptions underly-millions of well-characterized SNPs
[59,
62],
and rapid
ing their derivation
[77].
For these reasons, research
improvements in SNP genotyping technology have
groups often use simulation to empirically determine
ushered in the era of genomewide association studies
type I error (i.e. false-positive) rates in their own
(GWA), which utilize hundreds of thousands of SNPs
dataset, thereby assigning empirical significance levels
so that there is an increased chance of a genotyped
that may be evaluated for study-wide significance
[78].
marker being either: (i) a risk variant itself or (ii) close
enough to a risk variant so that they stay together
through many generations (i.e., in strong LD with
Association analysis
one another)
[91].
The first wave of GWA studies for
Whereas linkage identifies broad genetic regions (that
complex diseases has reported statistically significant
may contain hundreds of genes) linked with disease,
evidence for a number of variants. Strongly associated
association aims to identify a specific associated risk
SNPs have been identified and replicated for Crohn’s
allele. Association detects correlation between dis-Disease
[92, 93, 94],
type 1 diabetes
[93, 95],
type 2 dia-ease and marker allele(s)/haplotype(s) in a popula-betes
[93, 96, 97]
, and prostate cancer
[98];
for prostate
tion, which may be seen as one very large pedi-
cancer, three new studies
[99, 100, 101]
have replicated
Organic Syndromes of Schizophrenia – Section 3
multiple independent variants strongly associated
20 schizophrenia genome scans (1208 pedigrees, 2,945
with disease within a consensus linkage region on
affected), 12 additional studies have been reported in
chromosome 8q24
[98].
These findings highlight the
over 1,400 additional families from diverse popula-value of undertaking large-scale, complementary
tions, primarily supporting the linkages on 8p, 2q, 5p,
GWA studies across multiple populations
[98]
and
5q, 6p, 6q, and 10q.
underline the importance of common variants of
modest effect contributing to overall disease risk in
complex disease. The first published GWA for SZ
[102]
Biologically plausible SZ candidate genes have been
examined more than 500,000 markers in a case-control
identified primarily through their position in linkage
study of 178 SZ spectrum cases and reported a signifi-candidate regions or near cytogenetic abnormalities
cant association with SNP rs4129148 (p
=
3.7
×
10
−
7),
(Disrupted-in-Schizophrenia 1, DISC1, catechol-o-
near the colony stimulating factor receptor 2 alpha
methyltransferase, COMT). Additionally, candidates
(CSF2RA) gene in the pseudoautosomal region. This
have been identified through (i) differential gene
association surpassed the threshold for genomewide
expression in postmortem brain (e.g. regulator of
significance based on a Bayesian formula
[103].
This
G-protein signaling 4 (RGS4) gene, RGS4
[159]);
result awaits replication in larger samples.
(ii) psychopharmacological hypotheses regarding
Given the huge number of tests performed in large-functional polymorphisms in monoaminergic recep-
scale association studies, a critical issue is appropriate
tor genes (e.g. the dopamine receptor, DRD3
[160,
control of the type I error rate. A common approach
161];
5-hydroxytryptamine (serotonin) receptor 2A,
to multiple testing correction is Bonferroni correc-HTR2A
[162, 163]);
and (iii) specific hypotheses,
tion
[70,
104],
which assumes the independence of all
for example, aberrant signal transduction (RAC-tests performed. However, this approach is often overly
alpha serine/threonine-protein kinase AKT1,
[164].
conservative due to intermarker LD and nonindepen-
Table 23.1
lists SZ candidate genes currently attracting
dence of the individual tests. Other methods for deter-the most support. There have been a number of recent
mining statistical significance include the false discov-reviews of this literature
[56,
165, 166, 167, 168, 169,
ery rate and use of q-values
[105],
a Bayesian approach
170].
In this paper, relevant issues in SZ genetics will
involving the prior odds and power
[106],
and permu-be illustrated through an in-depth examination of one
tation methods including the application of extreme
candidate gene, DISC1.
value distributions to permuted data
[107].
DISC1: an illustrative example
After years of intensive international effort, linkage
Cytogenetics, linkage, and association
studies have achieved progress in identifying promis-
Figure 23.2
shows the genomic position, structure,
ing susceptibility regions for SZ. A number of regions
and published findings for DISC1 (and the neigh-
have demonstrated statistical significance in at least
boring gene, TSNAX). Jacobs originally reported
one study or strong support from multiple indepen-a chromosomal-balanced translocation disrupting
dent studies. These are shown in
Figure 23.1.
To
regions of chromosomes 1 and 11 (1;11)(q42;q14.3)
address the limitations of small samples, research
[190]
in the proband and other members of a four
groups have also combined their samples to con-
generational Scottish pedigree. Thirty-four of 77
duct large (> 700 SZ pedigrees), multicenter, region-pedigree members available for karyotyping car-
specific analyses that have provided support for some
ried this translocation
[191],
which generated a
regions including 6p and 8p
[108]
and 6q
[109]
but
LOD score of 7.1 with a disease phenotype crossing
no support for others such as 1q
[110]
and 22q
[111].
traditional diagnostic boundaries (including recur-Meta-analyses have highlighted somewhat different
rent major depression, bipolar disorder, and SZ);
but overlapping regions, with Badner and Gershon
the SZ LOD score was 3.6, and the LOD score for
[112]
supporting 8p, 13q, and 22q, and a rank-based
affective disorders was 4.5
[115].
The translocation
genome scan meta-analysis (GSMA)
[113]
supporting
directly disrupts the function of two novel genes on
2q plus a number of other regions including 3p, 8p,
1q42, termed Disrupted-In-Schizophrenia 1 and 2
and 22q. Since the GSMA was published on data from
(DISC1, DISC2)
[175].
DISC1 occupies
∼
415 Kb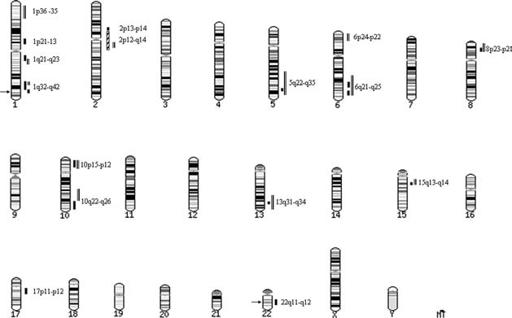
Chapter 23 – The status of genetic investigations of schizophrenia
Figure 23.1
Locations of linkage findings and chromosomal abnormalities implicated in SZ
Notes. Genomewide significant linkages (black line); consensus linkage regions attracting strong support from more than one independent
study (parallel lines); best region (2p12-q24) from rank based genome scan meta-analysis (GMSA)(striped box); arrows mark the sites of
chromosomal abnormalities: 1q42
[114, 115];
22q11:
[116, 117, 118, 119];