Ancient DNA: Methods and Protocols (27 page)
Read Ancient DNA: Methods and Protocols Online
Authors: Beth Shapiro
14. Adding EtBr to the running buffer offers the most dilute option, but increases the potential for splashing and also leads to EtBr contamination of the gel apparatus. Addition of EtBr to the gel prior to setting also contaminates the apparatus, but keeps the EtBr more contained than in the buffer. However, if EtBr is added to the gel, it must
never
be added before heating, as EtBr will be aerosolized, which is hazardous. Staining the gel after electrophoresis requires a more concentrated solution of EtBr than would be added to the running buffer, but provides a very contained region of contamination and often produces sharper DNA band images. Always dispose of EtBr-contaminated waste following your institution’s health and safety protocols.
15. Be sure that the samples are run slowly so that adequate separation occurs. As fragments are very small, it is important to obtain clear differentiation from primer-dimers, which are often not much smaller than the desired product.
16. If not all fragments are successfully amplifi ed, the same two-step multiplex procedure can be applied including only the primers for the missing fragments, or, in rare cases, single PCRs must be performed to achieve amplifi cation of all fragments.
Acknowledgments
The Pennsylvania State University and the National Science Foundation Award ANS-0909456 supported this work.
References
1. Hofreiter M, Jaenicke V, Serre D, Haeseler Av
multiplex DNA amplifi cation. Nucleic Acids
A, Pääbo S (2001) DNA sequences from mul—
Res 16:11141–11156
tiple amplifi cations reveal artifacts induced by 3. Edwards MC, Gibbs RA (1994) Multiplex cytosine deamination in ancient DNA. Nucleic
PCR: advantages, development, and applica—
Acids Res 29:4793–4799
tions. PCR Methods Appl 3:65–75
2. Chamberlain JS, Gibbs RA, Ranier JE, Nguyen
4. Hummel S, Schultes T, Bramanti B, Herrmann
PN, Caskey CT (1988) Deletion screening of
B (1999) Ancient DNA profi ling by megaplex
the Duchenne muscular dystrophy locus via
amplications. Electrophoresis 20:1717–1721
17 Multiplex PCR Amplifi cation of Ancient DNA
141
5. Schultes T, Hummel S, Herrmann B (1999)
Pinhasi R, Schmidt HA, Hofreiter M (2009)
Amplifi cation of Y-chromosomal STRs from
First DNA sequences from Asian cave bear
ancient skeletal material. Hum Genet
fossils reveal deep divergences and complex
104:164–166
phylogeographic patterns. Mol Ecol 18:
6. Krause J, Dear PH, Pollack JL, Slatkin M,
1225–1238
Spriggs H, Barnes I, Lister AM, Ebersberger I,
12. Stiller M, Baryshnikov G, Bocherens H,
Pääbo S, Hofreiter M (2006) Multiplex ampli—
Grandal d’Anglade A, Hilpert B, Münzel SC,
fi
cation of the mammoth mitochondrial
Pinhasi R, Rabeder G, Rosendahl W, Trinkaus
genome and the evolution of Elephantidae.
E, Hofreiter M, Knapp M (2010) Withering
Nature 439:724–727
away—25,000 years of genetic decline pre—
7. Römpler H, Dear PH, Krause J, Meyer M,
ceded cave bear extinction. Mol Biol Evol
Rohland N, Schöneberg T, Spriggs H, Stiller
27:975–978
M, Hofreiter M (2006) Multiplex amplifi ca—
13. Prost S, Knapp M, Flemmig J, Hufthammer
tion of ancient DNA. Nat Protoc 1:720–728
AK, Kosintsev P, Stiller M, Hofreiter M (2010)
8. Rohland N, Malaspinas AS, Pollack JL, Slatkin
A phantom extinction? New insights into
M, Matheus P, Hofreiter M (2008)
extinction dynamics of the Don-hare
Lepus
Proboscidean mitogenomics: chronology and
tanaiticus
. J Evol Biol 23:2022–2029
mode of elephant evolution using mastodon as
14. Römpler H, Rohland N, Lalueza-Fox C,
outgroup. PLoS Biol 5:e207
Willerslev E, Kuznetsova T, Rabeder G,
9. Richards MP, Pacher M, Stiller M, Quilès J,
Bertranpetit J, Schöneberg T, Hofreiter M
Hofreiter M, Constantin S, Zilhão J, Trinkaus E
(2006) Nuclear gene indicates coat-color poly-
(2008) Isotopic evidence for omnivory among
morphism in mammoths. Science 313:62
European cave bears: Late Pleistocene
Ursus
15. Campbell KL, Roberts JE, Watson LN,
spelaeus
from the Peştera cu Oase, Romania.
Stetefeld J, Sloan AM, Signore AV, Howatt JW,
Proc Natl Acad Sci USA 105:600–604
Tame JR, Rohland N, Shen TJ, Austin JJ,
10. Krause J, Unger T, Noçon A, Malaspinas AS,
Hofreiter M, Ho C, Weber RE, Cooper A
Kolokotronis SO, Stiller M, Soibelzon L,
(2010) Substitutions in woolly mammoth
Spriggs H, Dear PH, Briggs AW, Bray SC,
hemoglobin confer biochemical properties
O’Brien SJ, Rabeder G, Matheus P, Cooper A,
adaptive for cold tolerance. Nat Genet
Slatkin M, Pääbo S, Hofreiter M (2008)
42:536–540
Mitochondrial genomes reveal an explosive 16. Stiller M, Knapp M, Stenzel U, Hofreiter M, radiation of extinct and extant bears near the
Meyer M (2009) Direct multiplex sequenc—
Miocene-Pliocene boundary. BMC Evol Biol
ing (DMPS)—a novel method for targeted
8:220
high-throughput sequencing of ancient and
11. Knapp M, Rohland N, Weinstock J,
highly degraded DNA. Genome Res 19:
Baryshnikov G, Sher A, Nagel D, Rabeder G,
1843–1848
sdfsdf
Preparation of Next-Generation Sequencing Libraries
from Damaged DNA
Adrian W. Briggs and Patricia Heyn Abstract
Next-generation sequencing (NGS) has revolutionized ancient DNA research, especially when combined with high-throughput target enrichment methods. However, attaining high sequencing depth and accuracy from samples often remains problematic due to the damaged state of ancient DNA, in particular the extremely low copy number of ancient DNA and the abundance of uracil residues derived from cytosine deamination that lead to miscoding errors. It is therefore critical to use a highly effi cient procedure for conversion of a raw DNA extract into an adaptor-ligated sequencing library, and equally important to reduce errors from uracil residues. We present a protocol for NGS library preparation that allows highly effi cient conversion of DNA fragments into an adaptor-ligated form. The protocol incorporates an option to remove the vast majority of uracil miscoding lesions as part of the library preparation process. The procedure requires only two spin column purifi cation steps and no gel purifi cation or bead handling. Starting from an aliquot of DNA extract, a fi nished, highly amplifi ed library can be generated in 5 h, or under 3 h if uracil removal is not required.
Key words:
Ancient DNA
,
Ligation ,
Library preparation
,
Next-generation sequencing
,
High
throughput sequencing , Damage repair
1. Introduction
The development of next-generation sequencing (NGS) techniques has revolutionized genomics. By avoiding the capillary electrophoresis that limits the throughput of traditional Sanger sequencing, highly miniaturized and parallelized NGS platforms can perform tens of millions of sequence reactions per machine run, making it possible to generate whole eukaryote genome
sequences in a few days
( 1
) . The sequencing mechanisms and chemistries of current high-throughput platforms (Solexa-Illumina, 454-Roche, SOLiD-Applied Biosystems) are discussed elsewhere
( 2 )
, but all current high-throughput platforms use essentially the Beth Shapiro and Michael Hofreiter (eds.),
Ancient DNA: Methods and Protocols
, Methods in Molecular Biology, vol. 840, DOI 10.1007/978-1-61779-516-9_18, © Springer Science+Business Media, LLC 2012
143
144
A.W. Briggs and P. Heyn
same sample preparation principle. This principle is the construction of a sequencing library, where all DNA fragments in a sample of interest are fi rst end-repaired and then ligated to universal sequencing-adaptors. The universal adaptors allow amplifi cation and sequencing of all the DNA fragments in parallel using a single primer pair and sequencing primer.
In addition to the high raw sequence output, sequencing from adaptor-ligated libraries is extremely well suited to ancient DNA (aDNA) work for at least fi ve additional reasons: (1) the main drawback of NGS, short read lengths, is not a problem for aDNA since the DNA is already highly fragmented; (2) the use of adaptor primers outside the ancient fragments permits recovery of sequence information from molecules too short for conventional PCR, which is important since such fragments can constitute the vast majority of material in an ancient sample; (3) universal adaptors allow bulk PCR amplifi cation of an entire library before any downstream experiments, effectively immortalizing the library and greatly reducing the material demands on irreplaceable biological samples
( 3, 4 )
; (4) the 5’ and 3’ ends of fragments indicate unique DNA breakpoints and thus allow unique starting molecules to be counted at every genomic position
( 5
) , greatly increasing reliability and robustness against errors due to DNA damage or contamination; (5) library adaptors can be project-specifi cally barcoded, allowing downstream experiments to be performed outside the constrained environment of the aDNA cleanroom with no risk of fresh contamination by present day genomic DNA
( 6
) . Such downstream experiments often include targeted capture of genomic regions of interest prior to sequencing
( 4, 7, 8
) . Targeted capture avoids the high costs of sequencing irrelevant DNA from the sample, in particular the large amounts of microbial DNA present in most ancient remains.
The numerous advantages of aDNA sequencing by NGS
approaches have allowed for the fi rst time whole nuclear genomes to be sequenced from several thousand-year-old remains
( 9– 12
) , as well as long target regions from multiple individuals for population genetic analyses
( 4,
13, 14
) . Despite these successes, however, challenges remain when applying NGS to aDNA. After the death of an organism, its DNA is degraded by endogenous nucleases. DNA preserved over long time periods is also damaged by chemical and physical events, causing strand-breaks and base-modifi cations. Thus, aDNA is invariably highly fragmented and damaged
( 15
) . Although NGS libraries can be indefi nitely amplifi ed by adaptor-primed PCR, a major limiting factor in the NGS approach is the effi ciency with which aDNA molecules in the raw extract are successfully end-repaired and ligated to the adaptors during library preparation.
Clearly an ineffi cient library preparation protocol will strongly counteract the advantages of an NGS approach. In addition, base-18 Preparation of Next-Generation Sequencing Libraries from Damaged DNA 145
modifi cations are not repaired in standard NGS library preparation and can cause two problems: miscoding lesions and blocking lesions.
The major source for miscoding lesions in aDNA is deamination of cytosine to uracil, which gives rise to C–T transitions
( 16
) . It has been shown that uracil deamination is greatly increased in the regions close to the ends of fragments
( 6 )
, which are not analyzed in conventional PCR but form a substantial fraction of NGS data.
Base modifi cations that block DNA polymerase during amplifi cation remain poorly understood. However, recent evidence suggests that blocking lesions, if present, are at rather low frequency in aDNA and so may not be a major source of concern
( 17 )
.
Here we describe a NGS library preparation pr
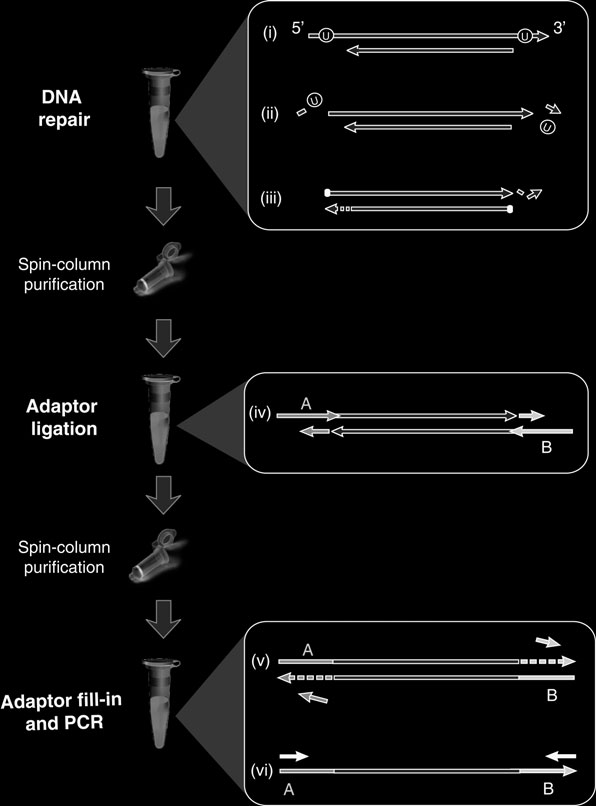
otocol (Fig. 1
) adapted to the requirements of aDNA work. In order to produce maximum library yield, the protocol employs an effi cient ligation procedure, involves only two spin column purifi cation steps before library amplifi cation, and does not require any bead handling steps.
An optional feature of the method is the inclusion of two repair enzymes (available pre-mixed from NEB), uracil-DNA-glycosylase and endonuclease VIII, that remove uracil and repair the DNA fragments afterwards, leaving the fragments amenable to sequencing yet free of miscoding lesions. This activity drastically reduces the transition error rate in fi nal sequences (at least 50-fold
( 18 )
), and therefore under most circumstances we strongly recommend using these enzymes. However, for certain applications the retention of miscoding errors in fi nal aDNA sequences may be desired, for example in studies of aDNA damage, or if damage itself will be used to assess the authenticity of pr
esumed aDNA ( 19, 20
) . In these cases the enzymes can be left out.
Our protocol allows conversion of a raw aDNA extract into a PCR-amplifi ed library in three to fi ve hours. The procedure is fully compatible with Roche/454, Illumina/Solexa or other NGS
sequencing platforms, simply requiring the appropriate sequences for the universal adaptors.
2. Materials
2.1. End Repair
1. 10× NEBuffer 2 (NEB, Ipswich, MA).
2. T4 polynucleotide kinase, 10 units/ m L (NEB, Ipswich, MA).
3. T4 DNA polymerase, 3 units/ m L (NEB, Ipswich, MA).
4. USER enzyme, 1 unit/ m L (NEB, Ipswich, MA).
2.2. Adaptor
1. Double-stranded library adaptors (2.5 mM) (see Note 1).
Attachment
2. Quick ligation kit (NEB, Ipswich, MA).
3. 3.dNTP mix (25 mM each of dATP, dTTP, dCTP, dGTP).