Ancient DNA: Methods and Protocols (41 page)
Read Ancient DNA: Methods and Protocols Online
Authors: Beth Shapiro
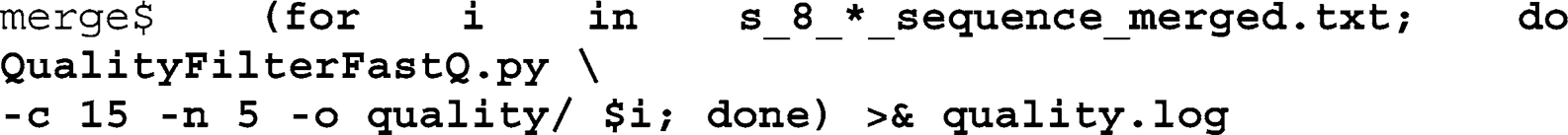
23 Analysis of High-Throughput Ancient DNA Sequencing Data 213
example, will only look at quality scores for prioritizing the search for non-perfect sequence matches. We therefore apply a simple, low stringency quality fi lter to our data that removes all reads that have more than fi ve bases with quality scores less than 15:
Iterate over the output fi les from the merging script and call the QualityFilterFastQ script with the quality threshold (-
c 15
) and the number of bases allowed below this threshold (-
n 5
), defi ne the output folder using the
o
-option, and redirect the command line output into a text fi le (
quality.log
):
This fi lter removed on average 4.8% of the reads for each of the 96 samples (minimum 1.4%, maximum 10.3%), and discarded
24.7% of the nonidentifi ed and low index quality sequences in “unknown.”
3.5. Identifi cation
One of the biggest challenges for aDNA is to distinguish endoge-
of Endogenous
nous sequences from those of environmental contaminants. In this
Molecules
situation, it is useful to know whether the potentially contaminating sequences have features that distinguish them easily from the endogenous DNA. GC content and length-based fi lters are typically insuffi cient to distinguish endogenous from exogenous DNA.
K
-mer frequencies, in particular the presence of longer
k
-mers, may be more useful (see Note 5).
Alignment approaches in which the sequence reads are aligned to the closest available reference sequence are often used. These may fail when the reference genome and the sample are evolution-arily distant, resulting in multiple substitutions or large indels. In this case, an overlap extension step (e.g., see ref.
( 56
) ) can be implemented, in which the unaligned reads (those that have not matched the reference genome) are used to attempt to extend the aligned reads (those that match the reference genome) into incompletely covered regions. Ideally, alignment and extension steps should be performed iteratively; however, there is no guarantee that such a process will converge. An iterative approach is also useful with
k
-mer fi lters, where
k
-mers identifi ed within all reads that pass a fi rst
k
-mer fi lter are used for an iterative identifi cation step. Alternatively, de Bruijn graphs (e.g.,
see
r
efs. ( 57– 60
) ) can be used to fi rst build contigs from overlapping
k
-mers. These contigs can then be fi ltered.
Alignments may outperform
k
-mer/de Bruijn graph approaches when samples are experimentally enriched for similarity to specifi c
214
M. Kircher
sequences, as the enrichment approaches will enrich for these same k -mers in the environmental DNA molecules.
Filters are generally applied in two ways: (1) negative/subtractive fi ltering, where reads identifi ed by some criterion are removed from the read set; and (2) positive fi ltering, where only reads identifi ed by some criterion are kept in the read set. Negative fi ltering may leave false sequences in the read set and remove highly conserved sequences, while positive fi ltering may miss divergent regions and include highly similar (e.g., conserved) false sequences.
The sequences in our example data set were enriched for their similarity to strepsirrhine primate mitochondrial genomes. We will therefore defi ne endogenous sequences by aligning the reads to any of the 14 complete mitochondrial genomes currently available from NCBI:
Create a FASTA fi le called
strepsirrhini.fasta
that contains these 14 sequences. Use the program BW
A ( 55 )
to perform a semi-global alignment (see Note 10) with gaps and substitutions, and store its output in BAM, the binary version of the Sequence Alignment/Map format
( 61
) . BWA expects either single read or paired-end data. Due to read merging and quality fi ltering, we have a mixture of both data types, and need to separate them prior to alignment. Use BWA with the parameters “ -l 65536 -n 0.01 -o 2” which will turn off seeding and allow more mismatches and two gap open events, rather than just a single gap (see Note 11).
Use the following two commands to generate index structures for BWA and samtools:
Next, we will separate single read and paired-end data (while discarding incomplete paired-end read pairs), perform alignment with BWA and convert the output to BAM:
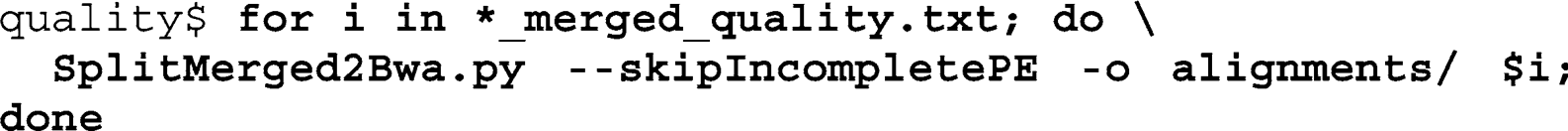
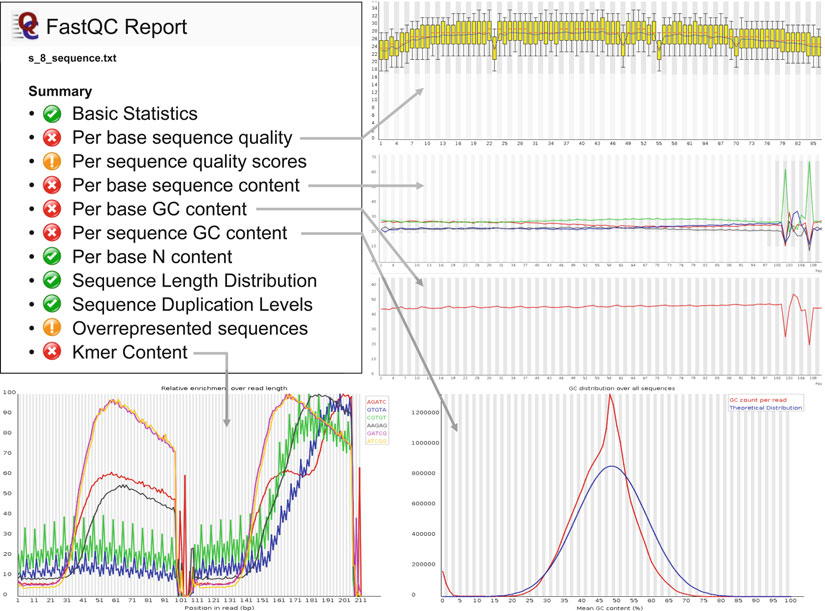
23 Analysis of High-Throughput Ancient DNA Sequencing Data 215
Fig. 6. Pieces of the HTML report for which FastQC reports an unexpected result. The fi rst three plots have been abbreviated to show only the fi rst half of the sequencing run (up to the end of the fi rst index). In the per base quality score plot, a few cycles show reduced average quality scores, but deviations are not extreme, and this is a reasonable result of quality score calibration. Per base sequence and GC content plots show spikes around the index reads, indicating an unbalanced base composition of the indexes selected. Further, per read GC content differs from the theoretical distribution. This may be caused by larger pieces of the adapters in the sequence reads as well as a GC bias from the enrichment process. The last fi gure shows overrepresentation of 5-mers over the read length. The high amplitude lines result from simple repeat sequences, which were also observed by TagDust and are an enrichment artifact. The other 5-mers are motifs from the adapter sequences and indicate early set-in of the library adapters, as is expected for short insert-size DNA.
First, create a new subfolder for the output fi les:
Use a for-loop to iterate over the quality-fi ltered fi les, calling the SplitMerged2Bwa script for each fi le. Exclude incomplete paired-end pairs generated by the quality fi lter, and write to the output folder created above:


216
M. Kircher
Change to the alignments folder:
Run BWA for the paired-end data and single read data separately, and then convert the paired-end alignments with BWA sampe to SAM and the single read data (the merged reads) with BWA samse to SAM. For the combined SAM output, use samtools to generate BAM, sorted by alignment coordinates. For further details, consult the samtools and BWA manuals.