Ancient DNA: Methods and Protocols (39 page)
Read Ancient DNA: Methods and Protocols Online
Authors: Beth Shapiro
TagDust requires a FastQ fi le with single read data. Below, use only the fi rst 50 nt of the forward read to identify artifacts. Generate the fi les using the TrimFastQ.py script . In addition, provide a list of the adapter sequences in a single FASTA fi le formatted as in the example fi le
double_multiplex_adapter.txt
below:




23 Analysis of High-Throughput Ancient DNA Sequencing Data 205
Run TrimFastQ.py, to create the input fi le with all sequences trimmed back to the fi rst 50 nt of the forward read:
Call TagDust, and request to write the identifi ed artifacts in FASTA format to a fi le called
artifacts.fa
. Provide the FASTA fi le with the adapter sequences (
double_multiplex_adapter.txt
) as well as the trimmed reads in FastQ:
Obtain the most frequent sequences in
artifacts.fa
using GNU
command line tools. The following command line ignores all lines in
artifacts.fa
that look like a FASTA header (i.e., start with “>”), sorts the remaining sequence lines, counts how many different sequences are obtained, then sorts them reverse numerical order and prints out the 30 most frequent sequences:
Now provide the same command as above, this time trimming
the sequences so that only the fi rst 30 nt of the identifi ed artifact sequences are considered:
Two distinct populations of sequences can be seen within the most frequent 30 sequences identifi ed by TagDust: one matching adapter dimer variants and the other matching mitochondrial
sequences (see Fig. 2 ). The second population likely r
esults from the enrichment process, which may alter the
k
-mer representation in the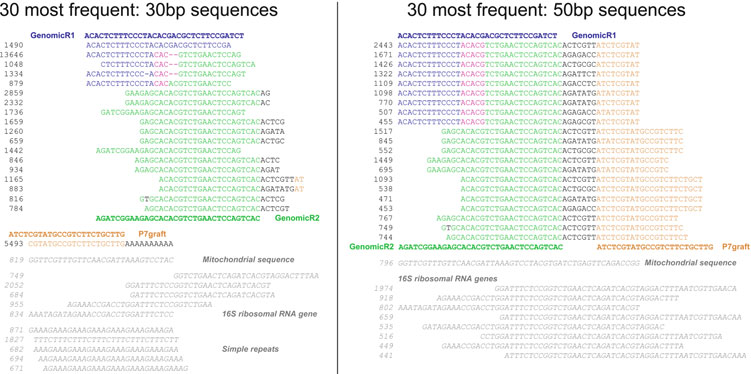
206
M. Kircher
Fig. 2. Adapter dimer variants and a population of false positive sequences (
italic
) that match the 30 most frequent artifact sequences reported by Ta
gDust ( 26 )
. Here, I consider only the fi rst 30 nt (
left
) or 50 nt (
right
) of the forward read for analysis.
samples and cause TagDust to identify as artifacts real sequences that happen to be present in high numbers. Thus, this direct application of computational fi ltering has its limits. In the next section, I will provide a protocol for fi ltering the identifi ed adapter dimer variants.
Other artifacts arising from the sequencing process are platform-specifi c and remain mostly unidentifi ed using the approach outlined here (see ref.
( 20 )
and see Note 6 for further details).
3.3. Adapter Trimming
Reads of short insert-size molecules often contain parts of the
and Merging
adapter sequence at the read end, which need to be identifi ed and
of Paired-End
removed so as not to interfere with downstream mapping or align-
Sequencing Data
ment. Unfortunately, this is not part of most data processing pipelines (with the exception of 454/Roche, see Note 7). This step is also nontrivial when sequencing error probability increases toward the end of reads.
Adapter identifi cation is simpler with paired reads than with single r
eads
( 8,
27, 28
) . For sequencing technologies without insertion/deletion errors, aligning the paired reads and identifying the overlapping region (autocorrelation) can reveal where the inser
t ends and adapter sequence begins (see Fig. 3 ). This appr
oach is more powerful than any alignment (-like) process for identifying adapters in single reads, as these will frequently remove short pieces of non-adapter sequences or miss adapter sequences due to higher sequencing error rates at the ends of reads.
Merging paired-end reads in short-insert libraries also decreases the number of sequencing err
ors ( 28
) . On a simulated Illumina data set with uniform read length distribution, the merging approach applied in refs.
( 8, 11, 13,
27, 28
) reduced the error rate of all merged sequences by a factor of about 5, and, for sequences shorter than or equal to the read length, by a factor of about 21
23 Analysis of High-Throughput Ancient DNA Sequencing Data 207
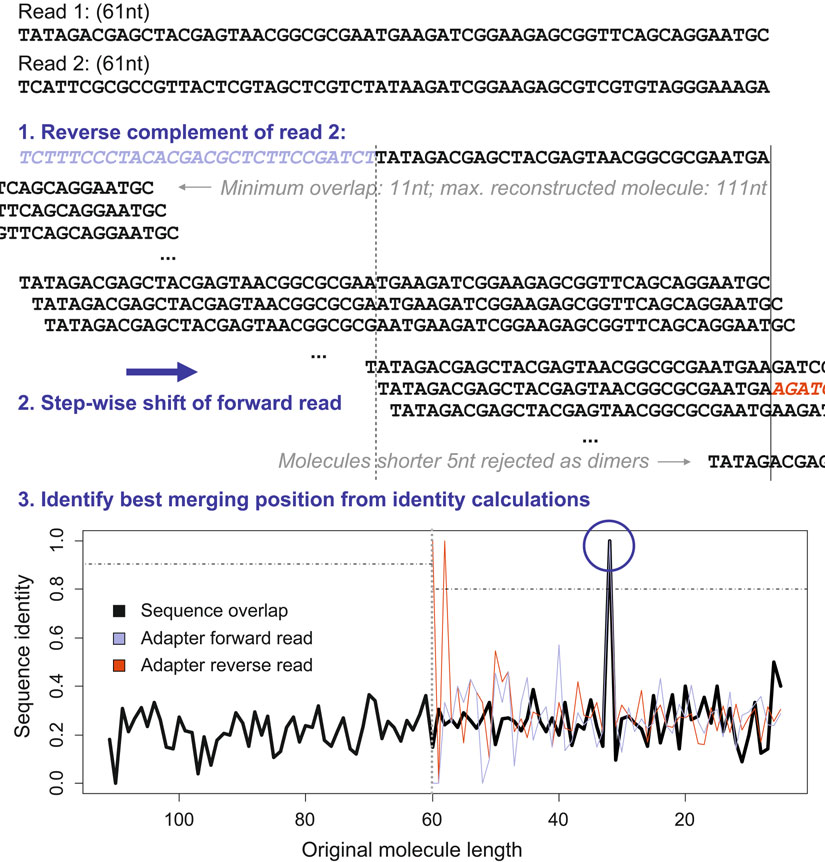
Fig. 3. Manipulating paired-end data. For paired-end data, identifying the adapter set-in point is simplifi ed by searching for overlapping sequence shared by the forward and reverse read. The fi gure illustrates how the forward read is shifted along the reverse complement of the second read to identify the original molecule length and fi nd the adapters (steps 1 and 2).
This is similar to the approach applied in refs.
( 8, 11, 13, 27, 28
)
, except: (1) the calculated sequence identity is corrected for the observed quality scores; and (2) a heuristic is implemented by fi rst searching the variants of decreasing length with adapter sequence present, and then checking the longer variants with no adapter sequence by increasing length. The search is aborted when a sequence identity of 95% is observed (step 3). If 95% identity is not observed, the maximum sequence identity is considered for read merging (reads are merged if at least 90% identity is observed when no adapter is present, or 80% identity is observed when at least one of the adapters is present). The implementation requires a minimum length of 11 nt for the overlap and rejects inserts shorter than 5 nt as adapter dimers.
(see Fig. 4
). Further, for sequences shorter than or equal to the read length, 99.997% of the simulated sequences were correctly merged. For the complete merging length range (5–191 nt) the original molecule length was recovered from 99.664% of reads.
The most frequent reason for the merge failure was sequencing 208
M. Kircher
Sequencing error in each read
0.3
Forward read
Reverse read
0
20
40
60
80
100
Position in read
0.2
Sequencing error [%]
0.1
Average of the two raw reads
Merged reads
0.0
0
50
100
150
Molecule size / library insert size
Fig. 4. Reduction in sequencing error rate caused by merging paired-end sequence data. Merging paired sequencing reads (
inset box
illustrates sequencing error of paired end reads) allows library adapters to be identifi ed and removed effi ciently and increases read accuracy. The
thick black line
shows the amount of sequencing error for different molecule lengths.
Note that as the length of the read increases, so does the sequencing error. Alternatively, the
thin line
shows the sequencing error that remains after application of the read-merging algorithm described in the main text. The data depicted are from a simulated data set of sequences ranging in length from 5 to 191 nt, generated with 2 × 101-cycles on an Illumina instrument with v4 chemistry. The data set was simulated with an error-informative quality score for which a random number (between 0 and 10, uniform sampling) was added to the average quality score for this sequence position when the correct base was simulated, and a random number subtracted if a wrong base was simulated.
errors in the overlapping regions (0.259%); false merging results are reported in only 0.077% of cases and mostly trace back to simple repeat sequences. Merging long insert libraries, where the overlapping region may be very small or not present, may cause incorrect reconstruction in particular of repeat regions. In this simulated data set, on average 0.29% of longer sequences (192–350 nt) were incorrectly reported as merged reads.
If paired-end data is not available, a requirement can be
imposed so that at least 5 nt of adapter sequence must be identifi ed if a sequence is to be included in downstream applications (false adapter identifi cation from 5 nt of random sequence is <0.1%). In addition, longer sequences (which may contain the adapter) can be excluded from all downstream analyses, so that only a minimal fraction of erroneous adapter sequences will be