Ancient DNA: Methods and Protocols (38 page)
Read Ancient DNA: Methods and Protocols Online
Authors: Beth Shapiro
py, FilterUniqueSAMCons.py,
SplitMerged2CDhit.py,
SplitFastQdoubleIndex.py, TrimFastQ.
py, MergeReadsFastQ_cc
samtools (v0.1.7a)
http://samtools.sourceforge.net
3. Methods
In many of the examples below, simple commands need to be typed into the command-line interface of your computer. In most cases, I fi rst provide descriptions, in words, of what the command is expected to achieve. I then provide (in bold face font) the actual commands that you should type in order to achieve the expected results.
3.1. Separation
Multiplexing and sample pooling are becoming more common in
of Individual Probes
HTS experiments. Barcoding (also called indexing or tagging) is
in Multiplex
often used when the target sequences comprise only a few loci or
Experiments
small genomes, and therefore sequencing only a single individual per lane or region would yield an excessive coverage. While sequencing platforms differ in how barcoded libraries are constructed, sequences from different libraries will be computationally sorted 23 Analysis of High-Throughput Ancient DNA Sequencing Data 201
post-sequencing via the index that is either part of the actual sequence read (index adjacent to insert) or sequenced as a separate technical read (index embedded in the adapter sequence). Typically, authors of the different barcoding protocols also provide software for separation by index (demultiplexing, e.g., see refs.
( 40– 43
) ) with the common result that the pooled sequences from a single run are written to separate fi les based on the identifi ed index sequence.
Demultiplex approaches differ in (1) whether only exact matches from a list of used/available index sequences are identifi ed or whether a limited number of errors is allowed, and (2) whether quality scores in the barcode read are used in this process. While using exact matches and requiring high quality scores provide the most conservative approach, this may not always be feasible. With long barcodes, for example, sequencing error may cause too many sequences to be excluded. Assuming a uniform error rate of 0.5% and a 6 nt index, around 3% of reads are predicted to contain errors. When barcode length is increased to 10 nt, around 5% of reads are expected to contain errors. Imbalanced use of bar
codes ( 40
) and the nonuniform distribution of errors across sequences
( 21, 25
) will increase the proportion of erroneous reads (~5–15%) with some sequence readouts showing close to random sequence. An intermediate solution is to apply a quality fi
lter (see Subheading 3.4 ) and to match a set of pr
e-compiled index variants containing very few substitutions.
Below, I provide an example workfl ow to separate sequences based on their index nucleotides. I use raw FastQ data from a 2 × 101-cycle Illumina paired-end sequencing lane (“s_8_sequence.txt”).
This data set contains a pool of 96 samples and an indexed φ X174
sequencing control library. All sample libraries have an average insert size of less than 200 nt. Half of the samples originate from ancient specimens while the other half are from modern specimens. In this experiment, indexed ancient and modern DNA samples have been pooled and used in an enrichment procedure (for mitochondrial sequences); therefore, the number of reads associated with each sample is expected to vary considerably. Samples are identifi ed by two, 7 nt indexes that are read in two technical reads: one after the forward read and the other after the reverse read. The FastQ fi le therefore contains sequences with a length of 216 nt: 101 nt forward read, 7 nt fi rst index, 101 nt reverse read, and 7 nt second index. The multiplex approach is an extension of the protocol described by Meyer and Kir
cher ( 40
) , in which the IS4 primer is replaced by a set of different index primers that introduce the second index r
ead ( 44 ). This setup is useful for identifying and excluding
experimental artifacts, such as index contamination or jumping PCR
( 45– 48
) that may occur during pooled library amplifi cation.
To create separate FastQ fi les for each index, use the
SplitFastQdoubleIndex Python script. In addition to the original FastQ fi le, provide the program with a three column, tab-separated text fi le with fi rst and second index sequence followed by the name
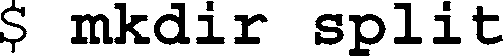
202
M. Kircher
of the sample (see Note 4). The fi le may contain one header line introduced by the hash character (“#”). For example:
When analyzing libraries created with either the original Meyer and Kir
cher ( 40
) protocol or similar pr
otocols ( 42, 49 )
, the same script can be used but with a two column fi le, leaving out the second index sequence. The script allows the user to choose whether to require perfect matches to each index or to allow mismatches between the sequence reads and the index used (see ref.
( 40 )
for details).
When using error correction, there can be either up to one mismatch or the loss of the fi rst base between the index that is read from the sequences and the “true” index sequence provided in the sample fi le. The script will create a separate output fi le for each of the defi ned index pairs and for index readouts that are found in the output FastQ
fi les but that have not been identifi ed by the user’s sample fi le (“unknown” indexes). In addition, a fi le will be created for index variants that cause two defi ned indexes to be less than two mismatches apart from each other (“confl ict” indexes), so that it is not possible to determine with certainty to which sample the index belongs. If index pairs are used for sample identifi cation and combinations of indexes encountered that are not defi ned by the user (incorrectly paired indexes), these reads are written into a “wrong” index fi le. Finally, valid index reads that have read quality scores below a provided threshold value can be automatically excluded. These quality-excluded sequences are saved in the “unknown” fi le with an asterisk (“*”) added to the FastQ read identifi er.
In the following example, run the demultiplex script on the example data set. First, using the command-line interface of your computer, create a new folder for the output fi les by typing the following command at the prompt:
Next, call the SplitFastQdoubleIndex script with the fi le containing the expected index pairs (
samples.tsv
). Defi ne the output folder and the start of the paired-end read in the FastQ input fi le (
s_8_sequence.txt
). Require a minimum quality score of 10 for the index reads and ask for a summary when the script fi nishes: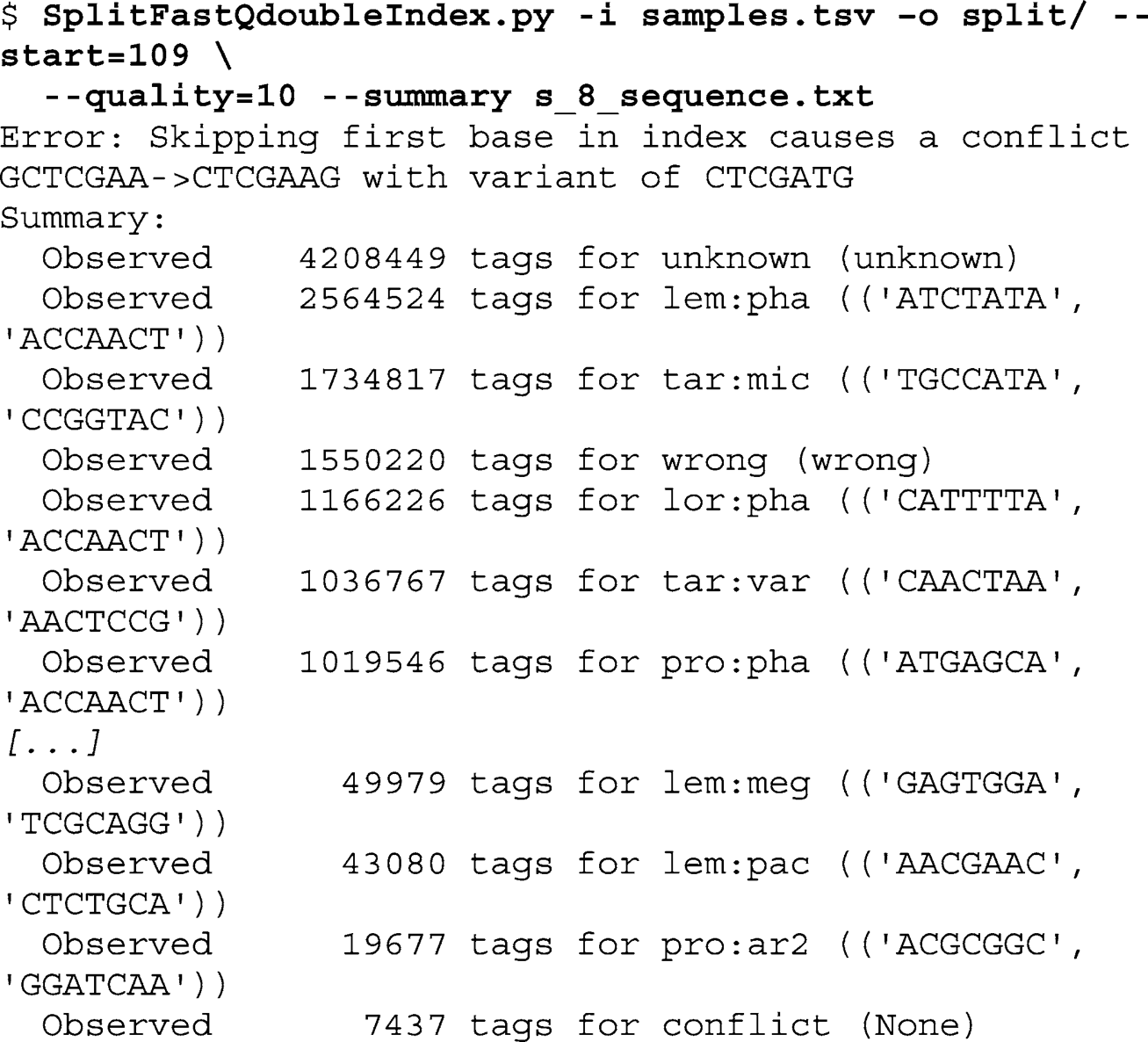

23 Analysis of High-Throughput Ancient DNA Sequencing Data 203
When changing to the output folder (split), you should now see 99 output fi les: one for each sample (96) plus the confl ict, unknown, and wrong fi les as described above. The new FastQ
sequence fi les no longer contain the sequence and quality scores of the index. Thus, sequences in the output fi les have a length 202 nt: 101 nt forward read and 101 nt reverse read.
Of the 47,584,117 sequences in
s_8_sequence.txt
, 4,208,449
(8.84%) are assigned to “unknown” indexes (931,550 or 1.96% of these were excluded by using a minimum quality score cutoff of 10).
Further, 1,550,220 (3.26%) sequences are “wrong” pairs and 7,437
are “confl ict” (0.02%; this is because two of the second read indexes
204
M. Kircher
are not at least two bases distant from each other if the loss of the fi rst base of the index is allowed). For the 96 samples used in this example, we obtain an average of 432,264 sequences per sample. As expected, the variation between samples is large: the best represented sample has 130 times more sequences than the least represented sample (minimum 19,677; maximum 2,564,524). In an ideal multiplex experiment, this factor should not exceed 10.
3.2. Removal
Most HTS platforms require platform-specifi c adaptors to be
of Protocol-Specifi c
ligated to the molecules in the DNA libraries prior to sequencing.
Library Artifacts
Library preparation protocols vary in their propensity to create library adapter dimers, chimeric sequences, and other artifacts that will need to be identifi ed and removed. In a typical HTS experiment using modern DNA, protocols are followed to enrich for molecules with correctly added adapters and to remove molecules with short or no inserts. When this is not possible, as is often the case with aDNA, library artifacts may dominate the resulting sequencing reads.
Programs such as T
agDust ( 26 )
compare the original adapter and primer oligonucleotide sequences with the output fi les to identify artifacts. The program can be used either to remove all sequence for which the library preparation oligonucleotide
k
-mers (see Note 5) make up the majority of the sequencing read (direct fi ltration) or to cluster the results from a single lane and identify the most frequently observed sequences, which can then be used for trimming and fi ltering with other softwar
e (see Subheading 3.3
). If TagDust is used to fi lter aDNA sequencing data, reads of short insert size (as may be common in degraded samples) may be excluded, simply because a large part of the sequence comprises library preparation oligonucleotides rather than the insert sequence. This could remove potentially informative sequences from the analysis.