Regenesis (12 page)
Authors: George M. Church
As I mentioned in
Chapter 2
, each amino acid typically bonds to one, two, or three of the thirty-two tRNA types, depending on the amino acid (as preparation for orderly assembly of them into long protein polymers). Any deviation is a serious error often resulting in the whole protein being junked by the cell (300 amino acids lost because of just a single wrongly positioned one!).
The mirror amino-acid-bonded tRNAs then constitute the “food” that a standard ribosome with standard mRNA can employ to make mirror proteins. Each tRNA is matched with mRNA, three base pairs at a time. The mRNA ratchets along through the ribosome (in units of these base triplets) directing the addition of further mirror amino acids to the growing protein. And at the end of the process,
voilà !
A mirror protein!
The anticodon from position 34 to 36, in this case GAA, binds with UUC. It can also bind to UUU, but binding to any other triplet would be a mistake and the ribosomal machinery has evolved to avoid this at all costs. The error rate of a ribosome reading triplets and adding the specified amino acid is less than 1 per 100,000. As it happens, the very first codon ever decoded was UUU, which coded for phenylalanine (F). In 1961 Marshall Nirenberg and Heinrich Matthaei noted that long stretches of UUU (poly-U) encode long polymers of F (poly-F). Moreover, the very first folded RNA gene product was the tRNA encoding F in 1977 (from the Rich, Kim, and Klug laboratories, assisted by a teenage version of myself). So, the molecules shown in
Figure 3.1
are historicâand also futuristic since the remarkable synthetic ribozyme pictured allows us to tear down the wall limiting us to the standard twenty or so amino acids in proteins ever since the code was formed at the dawn of life. All due to multiple forces maintaining the status quo, including metabolic enzymes, synthetases, and ribosomes that have evolved to destroy or avoid nonstandard amino acids.
To accomplish the transitional magic above (from standard to mirror amino acids), and to move beyond them to a fully mirror biological world, requires more work and more parts. Lots more work and parts. It requires more than just twenty tRNAs to accurately translate forty codons; you need thirty-two tRNAs to translate sixty-one codons, plus two release factor proteins to translate three stop codons; plus the ribosome consisting of fifty-four proteins and three RNAs, plus the initiation and elongation factors to start the translation and keep the mRNA ratcheting along. Oh, and we also need DNA and RNA polymerases, plus a few enzymes that modify the RNA, and proteins to make them more efficient and accurate. This project was briefly introduced in
Chapter 2
. All of these parts add up
to 113,000 base pairs, total, in the smallest minigenome capable of protein-based life, as Tony Forster and I laid out in a
Nature MSB
paper in 2006. This is
five times smaller
than the smallest free-living genome
(Mycoplasma)
. Every DNA, RNA, and protein molecule on this minigenome parts list would need to be chemically synthesized in a mirror version (all of the mirror monomers are already commercially available) or to be made by some cute transition trick like the one in
Figure 3.1
.
Just as life leads us to the enhancement of diversity, we now move from the question of how we achieve the minimal replexity capable of new functions, to the bigger questionâWhat are the maximum limits to (engineered) replexity? The above very compact minigenome will get us through the bottleneck of going from the RNA world to the RNA-plus-proteins world. But for most other purposes we want robust, complex metabolic and regulatory systems. This means that we need much more than the proteins required to make proteins from amino acids: we also need the protein enzymes to catalyze the synthesis of amino acid monomers from mineralsâand we also need thin air! The enzymes mentioned transform the atoms in sulfate and phosphate minerals, whereas the nitrogen and carbon dioxide of thin air are transformed into the atoms of amino acids and nucleotides. And as if that weren't enough, yet more proteins are needed to scavenge and horde resources, to wage war on other beings, and so on. Typically each does only one job well, so we need thousands of proteins per genome, and billions of proteins per ecosystem.
Ideally, for doing metabolic engineeringâmaking exotic biomaterials, pharmaceuticals, and chemicalsâwe would have all possible enzymes encoded in one master genome and we'd be able to turn their expression on and off, up and down, like a light switch, as needed for various tasks. So instead of a minimal genome, we want a
maximal
genomeâlike the vast online shopping world as opposed to the bare survival backpack. We call this collection of nearly all enzymes
E. pluri
, as in
E. coli
meets
E pluribus unum
. “From many states to one nation” becomes, in the world of genome
engineering, “From many enzyme genes to one integrated, customizable, all-purpose biofactory.”
While natural free-living cellular genomes range in size by a factor of a 150,000-fold differenceâfrom 580 kbp (
M.
genitalium
) to 90 billion bp (trumpet lily)âa rule of thumb is that a cell can double its genome size without any major reengineering. A cell can (and often does in the wild) double its own size, as well as double the size of its genome, without any help from human genome engineers. A cell can do this on its own by means of a variation on the process whereby a genome duplicates itself before the cell divides. If the cell wall misses its part of the division dance, the cell finds itself with twice the normal DNA content. This generally means a bigger cell that can be useful for intimidating or eating smaller cells. Later on it can be useful in providing vast resources for making new gene functions without hurting the old gene functions, since you now have two copies of each gene. When this is done by genome engineers they are simply taking advantage of the natural flexibility of cellular processes that have evolved across billions of years.
So,
E. pluri
might be able to accommodate 4,000 new genes encoding enzymes (each about 1 kbp long). To give a feeling for how huge this is, the full set of known enzymes not already in
E. coli
(and not counting minor variants) is 7,000. In much larger genomes the number of genes does not go up directly with total genomic bulk; this phenomenon is formally known as the C-value paradox, which states that the size of a genome does not correlate with the size or complexity of the organism, due to the presence of junk DNA. Possibly useful junk, but not all that useful.
It's an open question whether synthetic biology can overcome this limit to growth in complexity (1,000-fold increase in genome size from mustard to lily with no increase in gene number) if this is a trend in reality. As we head off into these complex cellular systems, we must reevaluate, for example, how to integrate multiple new amino acids into an already complex mixture. This integration requires freeing up some of the sixty-four codons (i.e., eliminating existing uses of those essential for life) and replacing them with orthogonal instances of bonding nonstandard amino
acids to tRNAs. (“Orthogonal” in this setting means highly reactive in making one kind of bond without getting engaged by any of the other bonds.)
The in vitro minigenome depends on the bonding of each tRNA with a specific amino acid using the simple 47-mer ribozyme depicted in
Figure 3.1
, but this process requires heavy human involvement in the lab and twenty separate reaction tubes. A normal cell has twenty synthetase enzymes, one for each of the twenty amino acids. Rarely does the wrong amino acid get bonded to a tRNA. The number of nonstandard amino acids (NSAAs) now exceeds seventy-five (largely from Peter Schultz and his students). The main barrier to expanding beyond the twenty standard amino acids is that all sixty-four codons already have an essential function and many have two such functions (e.g., overlaps of codons recognized by tRNAs; see
Figure 3.2
). For example, the anticodon for one of the tRNAs for valine (V) specifically binds to the mRNA codons GUU, GUA, GUG, and the other valine tRNA binds to GUU and GUC (in order to add a valine to the growing protein chain). So, even if we deleted the second tRNA, we would only free up GUC because GUU is still in use by the first tRNA.
The number of new synthetases (for bonding amino acids to the correct tRNAs) characterized so far is nine. These are intended to be specific for making only one kind of NSAA bond to one kind of new tRNA, with close to zero effect on any of the other NSAAs or standard twenty amino acids, but nothing is perfect. Even the standard twenty synthetases make mistakes at about 1 in 100,000 triesâand the use of all twenty-nine at once in the same cell is not yet fully tested. These
NSAAs
enable new chemical reactions specific to that NSAA without reacting with any other cellular molecules. Each new reaction chemistry was probably a really big deal evolutionarily (considering the amount of energy that, over the ages, nature devoted to evolving from four bases to twenty amino acids), and a big deal today too, as these chemistries will enable new routes to microbial drug manufacturing, new safety features in biotechnology, and so on.
But beyond RNA and proteins there's yet another innovation possible in the realm of protein or RNA backbones. Backbones are the constant
parts of the protein or RNA chemistry while the side chains (groups of molecules attached to the backbones) provide the variation (the spice of life). Normal (meaning naturally occurring) backbones consist of alternating sugar and phosphate molecules bonded together in an extended chain. Can we utilize totally new backbones? You bet! Another key polymer backbone is the polyketidesâpolymers of monomeric units consisting of two carbons plus one oxygen (and various derivatives of this) found in lipids and antibiotics.
Like RNA and proteins, the polyketide polymer is colinear with the basic information in DNA, meaning that the order of base pairs in the DNA determines the order of monomer type in the other polymers. The chemotherapeutic drug Doxorubicin is biosynthesized as a 10-mer (10 ketones in a linear polymer) with the sixth position modified from a ketone to an alcohol. In going from DNA to protein, the number of letters drops by threefold (e.g., UUU becomes F [phenylalanine]). In principle, we could make a ribosome that translates from mRNA directly to polyketides (instead of directly to polypeptides), but in nature this is accomplished in a more indirect manner by going from polynucleotide (mRNA) to polypeptide (enzyme) to polyketide. Each ketide monomer addition step requires its own enzyme domain (and these domains are each about 500 amino acids long), so the number of letters gets compressed down another 500-fold. This may seem verbose, but it gets the job done. Still, the possibility of constructing a more general and less verbose version on the ribosome is being explored in my lab.
Polyketides turn into long carbon chains (like fatty acids) and flat aromatic plates (like the tetracycline family of antibiotics). Long carbon chains and aromatics are the key components of diamonds and graphene, which have some truly amazing material propertiesâfor example, single-electron transistors, and/or the strongest and stiffest, have the highest thermal conductivity of any materials yet discovered (as recognized by the 2010 Nobel Prize for Chemistry). This class of biopolymer has many uses already when harvested from cells, and has even more profound applications in the future in biological and nonbiological systems as we get better at designing, selecting, and manufacturing this class of molecules.
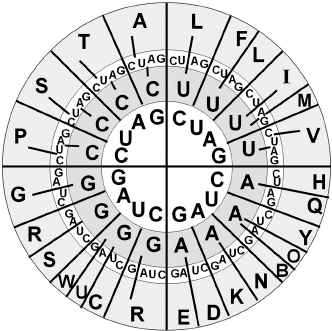
Figure 3.2
The translational code. Four RNA bases (U,C,A,G) make sixty-four triplets (3 base sequence in order from the innermost ring). 4*4*4 = 64. Each triplet encodes one of the twenty amino acids (they appear in the outermost ring: A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y). In addition, a few of the triplets encode three stop codons (UAA, UAG, UGA), which tell the ribosome to stop synthesizing whatever protein molecule is currently being made. These stop codons are often called by their historic names ochre, amber, and umber, and are abbreviated here B, O, and U, respectively. (O and U codons encode the less common amino acids pyrrolysine and selenocysteine, respectively, in some organisms.) In the early 1960s, members of the Steinberg, Epstein, and Benzer labs at Caltech were studying viruses that infect some strains of
E. coli
and not others. These researchers offered to name any mutant that might have these amazing properties after the one who found it. It just so happened that a student named Harris Bernstein (who normally worked on fungi, not viruses) found it. “Bernstein” in German means “amber,” and so they named the UAG stop codon amber. They later dubbed the other two stop codons ochre and umber to maintain the color theme. To let everyone know how hip you are, you can purchase (at
thednastore.com
) a bumper sticker that says “I Stop for UAA.”