In Search of Memory: The Emergence of a New Science of Mind (27 page)
Read In Search of Memory: The Emergence of a New Science of Mind Online
Authors: Eric R. Kandel
Tags: #Psychology, #Cognitive Psychology & Cognition, #Cognitive Psychology
15–1 Anatomical changes accompany long-term memory
.
Thus in
Aplysia
we could see for the first time that the number of synapses in the brain is not fixed—it changes with learning! Moreover, long-term memory persists for as long as the anatomical changes are maintained.
These studies provided the first clear insights into the two competing theories of memory storage. Both were right, but in different ways. Consistent with the one-process theory, the same site can give rise to both short-term and long-term memory in habituation and sensitization. Moreover, in each case a change in synaptic strength occurs. But consistent with the two-process theory, the mechanisms of short- and long-term change are fundamentally different. Short-term memory produces a change in the function of the synapse, strengthening or weakening preexisting connections; long-term memory requires anatomical changes. Repeated sensitization training (practice) causes neurons to grow new terminals, giving rise to long-term memory, whereas habituation causes neurons to retract existing terminals. Thus, by producing profound structural changes, learning can make inactive synapses active or active synapses inactive.
TO BE USEFUL, A MEMORY HAS TO BE RECALLED. MEMORY
retrieval depends on the presence of appropriate cues that an animal can associate with its learning experiences. The cues can be external, such as a sensory stimulus in habituation, sensitization, and classical conditioning, or internal, sparked by an idea or an urge. In the
Aplysia
gill-withdrawal reflex, the cue for memory recall is external: namely, the touch to the siphon that elicits the reflex. The neurons that retrieve the memory of the stimulus are the same sensory and motor neurons that were activated in the first place. But because the strength and number of synaptic connections between these neurons have been altered by learning, the action potential generated by the sensory stimulus to the siphon “reads out” the new state of the synapse when it arrives at the presynaptic terminals and the recall gives rise to a more powerful response.
In long-term memory, as in short-term memory, the number of changed synaptic connections may be great enough to reconfigure a neural circuit, but this time anatomically. For example, prior to training, a stimulus to a sensory neuron in
Aplysia
might be strong enough to cause motor neurons leading to the gill to fire action potentials, but not strong enough to cause motor neurons leading to the ink gland to fire action potentials. Training strengthens not only the synapses between the sensory neuron and the motor neurons to the gill but also the synapses between the sensory neuron and the motor neurons to the ink gland. When the sensory neuron is stimulated after training, it retrieves the memory of the enhanced response, which causes both gill and ink motor neurons to fire action potentials and causes inking as well as gill withdrawal to take place. Thus, the form of
Aplysia
’s behavior is altered. The touch to the siphon elicits not just a change in the magnitude of the behavior—the amplitude of gill withdrawal—but also a change in the animal’s behavioral repertory.
Our studies showing that the brain of
Aplysia
is physically changed by experience led us to wonder, Does experience change the primate brain? Does it change the brains of people?
WHEN I WAS A MEDICAL STUDENT IN THE 1950S, WE WERE TAUGHT
that the map of the somatosensory cortex discovered by Wade Marshall is fixed and immutable throughout life. We now know that idea is not correct. The map is subject to constant modification on the basis of experience. Two studies in the 1990s were particularly informative in this regard.
First, Michael Merzenich at the University of California, San Francisco discovered that the details of cortical maps vary considerably among individual monkeys. For example, some monkeys have a much more extensive representation of the hand than other monkeys. Merzenich’s initial study did not separate the effects of experience from those of genetic endowment, so it was possible that the differences in representation were genetically determined.
Merzenich then carried out additional experiments to determine the relative contributions of genes and experience. He trained monkeys to obtain food pellets by touching a rotating disk with their three middle fingers. After several months, the area of the cortex devoted to the middle fingers—especially the tips of the fingers used for touching the disk—had expanded greatly (figure 15–2). At the same time, the tactile sensitivity of the middle fingers increased. Other studies have shown that training in visual discrimination of color or form also leads to changes in brain anatomy and improved perceptual skills.
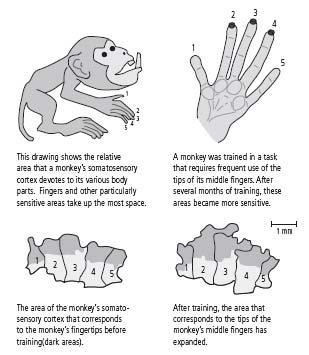
15–2 Maps of the cortex change with experience.
(Adapted from Jenkins et al., 1990.)
Second, Thomas Ebert and his colleagues at the University of Konstanz in Germany compared images of violinists’ and cellists’ brains with images of nonmusicians’ brains. Players of stringed instruments use the four fingers of the left hand to modulate the sound of the strings. The fingers of the right hand, which move the bow, are not involved in such highly differentiated movements. Ebert found that the area of the cortex devoted to the fingers of the right hand did not differ in string players and nonmusicians, whereas representations of the fingers of the left hand were much more extensive—by as much as five times—in the brains of string players than in those of nonmusicians. Furthermore, musicians who began playing the instrument before age thirteen had larger representations of the fingers of their left hand than musicians who began playing after that age.
These dramatic changes in cortical maps as a result of learning extended the anatomical insights that our studies in
Aplysia
had revealed: the extent to which a body part is represented in the cortex depends on the intensity and complexity of its use. In addition, as Ebert’s study showed, such structural changes in the brain are more readily achieved in the early years of life. Thus, a great musician such as Wolfgang Amadeus Mozart is who he is not simply because he has the right genes (although genes help), but also because he began practicing the skills for which he became famous at a time when his brain was more pliable.
Moreover, our results in
Aplysia
showed that the plasticity of the nervous system—the ability of nerve cells to change the strength and even the number of synapses—is the mechanism underlying learning and long-term memory. As a result, because each human being is brought up in a different environment and has different experiences, the architecture of each person’s brain is unique. Even identical twins with identical genes have different brains because of their different life experiences. Thus, a principle of cell biology that first emerged from the study of a simple snail turned out to be a profound contributor to the biological basis of human individuality.
Our finding that short-term memory results from a functional change and long-term memory from an anatomical change raised even more questions: What is the nature of memory consolidation? Why does it require the synthesis of new protein? To find out, we would have to move into the cell and study its molecular makeup. My colleagues and I were ready for that step.
JUST AT THIS POINT, WE HEARD DEVASTATING NEWS. IN THE FALL
of 1973 Alden Spencer, my best friend and the cofounder of the Neurobiology and Behavior Division at NYU, began complaining of weakness in his hands, which interfered with his tennis game. Within a few months he was diagnosed as having amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease), a disease that is invariably fatal. On hearing this diagnosis from one of the country’s leading neurologists, Alden became depressed and began preparing his will, thinking he might be dead within a week. But Alden also had arthritis of the elbow, a feature not typically associated with ALS. I therefore suggested that he see a rheumatologist.
Alden saw a very good doctor who assured him that he did not have ALS but instead had a connective tissue disorder (a collagen disease) related to lupus erythematosus. On hearing this much more optimistic diagnosis, Alden’s mood improved. A few months later he went back to his neurologist, who assured him that independent of any arthritis, he clearly had ALS. Alden’s mood immediately fell again.
At that point I spoke to the neurologist and told him that Alden was clearly having great difficulty handling his diagnosis, and I asked the neurologist whether he could help Alden by holding out more hope. The neurologist, a thoroughly decent and caring person, insisted that he could not do that because it would deceive Alden about his future, which would not be fair to him. “But,” he said, “I have nothing to offer Alden. He simply need not and should not come to see me. Let him continue to see the rheumatologist.”
I discussed this course with Alden and independently with his wife, Diane. Both thought it a good idea. Diane was convinced that Alden did not want to confront what she and I had sadly come to agree must be the correct diagnosis, that he had ALS.
During the next two and a half years, Alden slipped slowly and progressively. He first used a cane and then a wheelchair to get around. But at no time did he stop going to his laboratory and doing science. Even though giving lectures became difficult for him, he continued to teach, albeit fewer classes. No one in our group except me knew his true diagnosis, and no one thought, or at least acknowledged, that he did not have a peculiar form of arthritis. He exercised continually and swam on a regular basis at a special swimming pool for the disabled near his house. The day before he died, in November 1977, he was in his laboratory participating in a discussion on sensory processing.
Alden’s death was shattering to all of us personally and devastating to our close-knit group. We had talked almost daily for about twenty years, so for a long while afterward the whole rhythm of my working life was disrupted. I still think of Alden frequently.
I was not alone; everyone enjoyed Alden’s self-deprecating humor, his modesty and unbounded generosity, and his unending creativity. To honor his memory, we established in 1978 the Alden Spencer Lectureship and Award, given annually to a great scientist under the age of fifty whose best work is still ahead of him or her. The recipient is selected by the entire Center for Neurobiology and Behavior at Columbia—faculty, graduate students, postdoctoral fellows, and professors.
The years following Alden’s death were productive and therefore seemed harmonious from the outside, but they were very painful for me personally. Alden’s death in 1977 was followed by my father’s death the same year and my brother’s death in 1981. In each case I was extensively involved in their care, and their deaths left me not only psychologically despondent and depleted but also physically exhausted. I have always been grateful for the serenity I have been able to obtain by focusing hard on my work. At that time, the challenging quest of my work and the surprising insights revealed to me were a particularly welcome retreat from the painful realities of everyday life’s irretrievable losses.
This difficult period was made even more painful for me by my son Paul’s departure for college in 1979. When Paul was seven years old, I encouraged him to take up chess and to take lessons in tennis, and he subsequently became quite good at both. I played chess and therefore engaged his interest in rooks and knights and checkmates. But I did not play tennis. So at age thirty-nine, I started to take tennis lessons and soon had a mediocre but highly enjoyable game, which I still play regularly. From the time Paul first began playing tennis, he was one of my regular partners. By his last year of high school, he had developed into a wonderfully good player and was my only partner. His leaving home deprived me not just of my son, but also of my tennis and chess partner. I was beginning to feel like Job.
I
n 1975, twenty years after Harry Grundfest told me that we needed to study the brain one cell at a time, my colleagues and I had begun to explore the cellular basis of memory—how one can remember a social encounter, a natural scene, a lecture, or a medical pronouncement for a lifetime. We had learned that memory derives from changes in the synapses in a neural circuit: short-term memory from functional changes and long-term memory from structural changes. Now we wanted to delve even deeper into the mystery of memory. We wanted to penetrate the molecular biology of a mental process, to know exactly what molecules are responsible for short-term memory. With that question, we were entering completely uncharted territory.
THE JOURNEY WAS MADE LESS DAUNTING BY MY INCREASING
confidence that in
Aplysia
we had a simple system in which we could explore the molecular basis of memory storage. We had entered the labyrinth of synaptic connections in
Aplysia
’s nervous system, mapped the neural pathway of its gill-withdrawal reflex, and shown that the synapses forming it could be strengthened by learning. We had quite literally navigated the outer rings of a scientific maze. Now we wanted to determine exactly where along this neural pathway the synaptic changes associated with short-term memory are localized.
WE FOCUSED OUR ATTENTION ON THE CRITICAL SYNAPSE BETWEEN
the sensory neuron that transmits information about touch from the snail’s siphon and the motor neuron whose action potentials cause the gill to withdraw. We now wanted to know how the two neurons that make up the synapse contribute to the learned change in synaptic strength. Does the sensory neuron change in response to the stimulus, leading its axon terminals to release more or less transmitter? Or does the change occur in the motor neuron, resulting in an increased number of receptors in the cell to the neurotransmitter or an increase in the sensitivity of receptors to the transmitter? We found that the change is quite one-sided: during short-term habituation lasting minutes, the sensory neuron releases less neurotransmitter, and during short-term sensitization it releases more neurotransmitter.
That neurotransmitter, we later discovered, is glutamate, also the major excitatory transmitter in the mammalian brain. By increasing the amount of glutamate a sensory cell sends to a motor cell, sensitization strengthens the synaptic potential elicited in the motor cell, thus making it easier for that neuron to fire an action potential and cause the gill to withdraw.
The synaptic potential between the sensory and motor neurons lasts only milliseconds, yet we had observed that a shock to
Aplysia
’s tail enhances glutamate release and synaptic transmission for many minutes. How does this come about? As my colleagues and I focused on the question, we noticed something curious. The strengthening of the synaptic connection between the sensory and motor neuron is accompanied by a very slow synaptic potential in the sensory cell, one that lasts for minutes rather than the milliseconds typical of synaptic potentials in the motor neuron. We soon found that the shock to
Aplysia
’s tail activates a second class of sensory neurons, one that receives information from the tail. These tail sensory neurons activate a group of interneurons that acts on the sensory neuron from the siphon. It is these interneurons that produce the remarkably slow synaptic potential. We then asked ourselves: What neurotransmitter do the interneurons release? How does this second neurotransmitter lead to the release of more glutamate from the terminals of the sensory neuron, thus creating short-term memory storage?
We found that the interneurons activated by a shock to
Aplysia
’s tail release a neurotransmitter called serotonin. Moreover, the interneurons form synapses not only on the cell body of the sensory neurons but also on the presynaptic terminals, and they not only produce a slow synaptic potential but also enhance the sensory cell’s release of glutamate onto the motor cell. In fact, we could simulate the slow synaptic potential, the enhancement of synaptic strength, and the strengthening of the gill-withdrawal reflex simply by applying serotonin to the connections between the sensory and motor neurons.
We called these serotonin-releasing interneurons modulatory interneurons because they do not mediate behavior directly; rather, they modify the strength of the gill-withdrawal reflex by enhancing the strength of the connections between sensory and motor neurons.
These findings caused us to realize that there are two kinds of neural circuits important in behavior and learning: mediating circuits, which we had characterized earlier, and modulating circuits, which we were just beginning to characterize in detail (figure 16–1). Mediating circuits produce behavior directly and are therefore Kantian in nature. These are the genetically and developmentally determined neuronal components of the behavior, the neuronal architecture. The mediating circuit is made up of the sensory neurons that innervate the siphon, the interneurons, and the motor neurons that control the gill-withdrawal reflex. With learning, the mediating circuit becomes the student and acquires new knowledge. The modulating circuit is Lockean in nature; it serves as a teacher. It is not directly involved in producing a behavior but instead fine-tunes the behavior in response to learning by modulating—heterosynaptically—the strength of synaptic connections between the sensory and motor neurons. Activated by a shock to the tail, a completely different part of the body than the siphon, the modulating circuit teaches
Aplysia
to pay attention to a stimulus to the siphon that is important for its safety. Thus the circuit is, in essence, responsible for arousal or salience in
Aplysia
, just as analogous modulatory circuits are an essential component of memory in more complex animals, as we will see later.
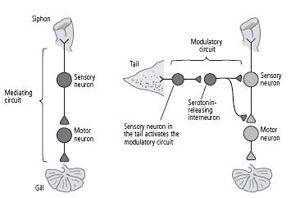
16–1 The two types of circuits in the brain.
Mediating circuits produce behaviors. Modulatory circuits act on the mediating circuits, regulating the strength of their synaptic connections.
That serotonin was a modulator for sensitization simply amazed me! Some of my first experiments with Dom Purpura in 1956 had focused on the action of serotonin. In fact, on Student Day at NYU Medical School in the spring of 1956, I had given a brief talk entitled “Electrophysiological Patterns of Serotonin and LSD Interaction on Afferent Cortical Pathways.” Jimmy Schwartz had been kind enough to listen to a rehearsal of the talk and to help me improve it. I was now beginning to appreciate that life is circular. I had not worked on serotonin for almost twenty years, and here I was returning to it with renewed focus and enthusiasm.
ONCE WE KNEW THAT SEROTONIN ACTED AS A MODULATORY
transmitter to enhance the release of glutamate from the presynaptic terminals of the sensory neuron, the stage was set for a biochemical analysis of memory storage. Fortunately, in Jimmy Schwartz I had an excellent guide and fellow traveler on this journey.
Before returning to NYU, Jimmy had worked at Rockefeller University on the bacterium
Escherichia coli
, the single-celled organism in which many fundamental principles of modern biochemistry and molecular biology were first worked out. In 1966 his interest had shifted to
Aplysia
, and he began his research by delineating the chemical transmitters used by a neuron in the abdominal ganglion. In 1971, we joined forces to study the molecular actions that accompany learning.
Jimmy was of inestimable help in this second major stage of my biological education. We were influenced by the work of Louis Flexner, who had shown a few years earlier that long-term memory in mice and rats requires the synthesis of new protein, whereas short-term memory does not. Proteins are the workhorses of the cell. They constitute its enzymes, ion channels, receptors, and transport machinery. Since, as we had found, long-term memory involves the growth of new connections, it is not surprising that the synthesis of new protein constituents is required for that growth.
Jimmy and I set out to test this idea in
Aplysia
and to do so at the level of the siphon sensory cell and its synapses on the motor neurons to the gill. If synaptic changes parallel changes in memory, then the short-term synaptic changes we had delineated should not require the synthesis of new protein. That is exactly what we found. What, then, mediates this short-term change?
Cajal had shown that the brain is an organ constructed of neurons wired to each other in specific pathways. I had seen this remarkable connection specificity in the simple neural circuits that mediate reflex behavior in
Aplysia
. But Jimmy pointed out that this specificity also extends to molecules—to the combinations of atoms that serve as the elementary units of cellular function. Biochemists had found that molecules can interact with one another within a cell and that these chemical reactions are organized in specific sequences known as biochemical signaling pathways. The pathways convey information in the form of molecules from the surface of the cell to the interior, much as one nerve cell conveys information to another. In addition, the pathways are “wireless.” Molecules floating within the cell recognize and bind to specific molecular partners and regulate their activity.
My colleagues and I had not only fulfilled my early ambition of trapping a learned response in the smallest possible population of neurons, we had trapped a component of a simple form of memory in a single sensory cell. But even a single
Aplysia
neuron contains thousands of different proteins and other molecules. Which of these molecules are responsible for short-term memory? As Jimmy and I began to discuss the possibilities, we focused in on the idea that the serotonin released in response to a shock to the tail might enhance glutamate release from the sensory neuron by launching a specific sequence of biochemical reactions in the sensory cell.
The sequence of biochemical reactions that Jimmy and I sought would have to serve two fundamental purposes. First, they would have to translate the brief action of serotonin into molecules whose signals would last for minutes within the sensory neuron. Second, those molecules would have to broadcast signals from the cell membrane, where serotonin acts, to the interior of the sensory cell, particularly to the specialized regions of the axon terminal involved in the release of glutamate. We elaborated on these thoughts in our 1971 article in the
Journal of Neurophysiology
and speculated on the possibility that a specific molecule known as cyclic AMP might be involved.
WHAT IS CYCLIC AMP? HOW DID WE HIT UPON IT AS A LIKELY
candidate? Cyclic AMP came to mind because this small molecule was known to serve as a master regulator of signaling within muscle and fat cells. Jimmy and I knew that nature is conservative—therefore, a mechanism used in the cells of one tissue is likely to be retained and used in the cells of another tissue. Earl Sutherland at Case Western Reserve University in Cleveland had already shown that the hormone epinephrine (adrenaline) produces a brief biochemical change at the surface membrane of fat and muscle cells that gives rise to a more enduring change inside the cells. That longer lasting change is brought about by an increase in the amount of cyclic AMP inside those cells.
Sutherland’s revolutionary findings came to be described as the second-messenger signaling theory. The key to this biochemical signaling theory was his discovery of a new class of receptors at the cell surface of fat and muscle cells that responds to hormones. Earlier, Bernard Katz had found the neurotransmitter-gated receptors known as ionotropic receptors; on binding a neurotransmitter, these receptors open or close the gate of an ion channel contained within the receptor, thus translating a chemical signal into an electrical signal. But the new class of receptors, called metabotropic receptors, has no ion channel within it to open or close. Instead, one region of these receptors protrudes from the outside surface of the cell membrane and recognizes signals from other cells, while another region protrudes from the inside of the cell membrane and engages an enzyme. When these receptors recognize and bind a chemical messenger on the outside of the cell, they activate an enzyme within the cell called adenylyl cyclase, which makes cyclic AMP.
This process has the advantage of having greatly amplifying the cell’s response. When one molecule of chemical messenger binds to a metabotropic receptor, that receptor stimulates adenylyl cyclase to make a thousand molecules of cyclic AMP. Cyclic AMP then binds to key proteins that trigger a whole family of molecular responses throughout the cell. Finally, adenylyl cyclase continues making cyclic AMP for minutes. The actions of metabotropic receptors therefore tend to be more powerful, more widespread, and more persistent than the actions of ionotropic receptors. Whereas ionotropic actions typically last milliseconds, metabotropic actions can last from seconds to minutes—one thousand to ten thousand times longer.